The State of Greenhouse Gases in the Atmosphere Using Global Observations through 2017
On-line version of the WMO Greenhouse Gas Bulletin N.14
Executive summary
The latest analysis of observations from the WMO GAW Programme shows that globally averaged surface mole fractions calculated from this in situ network for carbon dioxide (CO2), methane (CH4) and nitrous oxide (N2O) reached new highs in 2017, with CO2 at 405.5 ± 0.1 ppm(2), CH4 at 1859 ± 2 ppb and N2O at 329.9 ± 0.1 ppb. These values constitute, respectively, 146%, 257% and 122% of pre-industrial (before 1750) levels. The increase in CO2 from 2016 to 2017 was smaller than that observed from 2015 to 2016 and practically equal to the average growth rate over the last decade. The influence of the El Niño event that peaked in 2015 and 2016 and contributed to the increased growth rate during that period sharply declined in 2017. For CH4, the increase from 2016 to 2017 was lower than that observed from 2015 to 2016 but practically equal to the average over the last decade. For N2O, the increase from 2016 to 2017 was higher than that observed from 2015 to 2016 and practically equal to the average growth rate over the past 10 years. The NOAA Annual Greenhouse Gas Index (AGGI) [4] shows that from 1990 to 2017 radiative forcing by long-lived GHGs (LLGHGs) increased by 41%, with CO2 accounting for about 82% of this increase.
|
Overview
This fourteenth WMO Greenhouse Gas Bulletin reports atmospheric abundances and rates of change of the most important LLGHGs – CO2, CH4 and N2O – and provides a summary of the contributions of other gases. These three, together with CFC-12 and CFC-11, account for approximately 96% of radiative forcing due to LLGHGs (Figure 1).
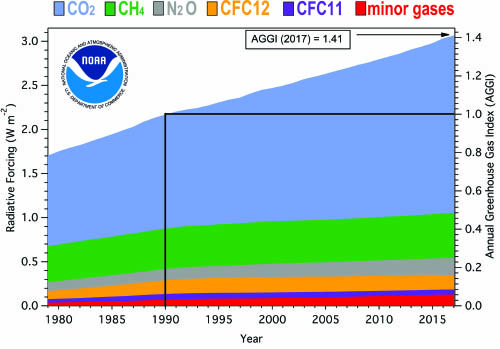
Figure 1.
Atmospheric radiative forcing, relative to 1750, of LLGHGs and the 2016 update of the NOAA AGGI. |
|
The GAW Programme (http://www.wmo.int/gaw) coordinates systematic observations and analysis of GHGs and other trace species. Sites where GHGs have been measured in the last decade are shown in Figure 2. Measurement data are reported by participating countries and archived and distributed by the WMO World Data Centre for Greenhouse Gases (WDCGG) at the Japan Meteorological Agency.
The results reported here by WDCGG for the global average and growth rate are slightly different from results reported by NOAA for the same years [6] due to differences in the stations used, differences in the averaging procedure and a slightly different time period for which the numbers are representative. WDCGG follows the procedure described in detail in [7].
Unexpected Increases in Global Emissions of CFC - 11
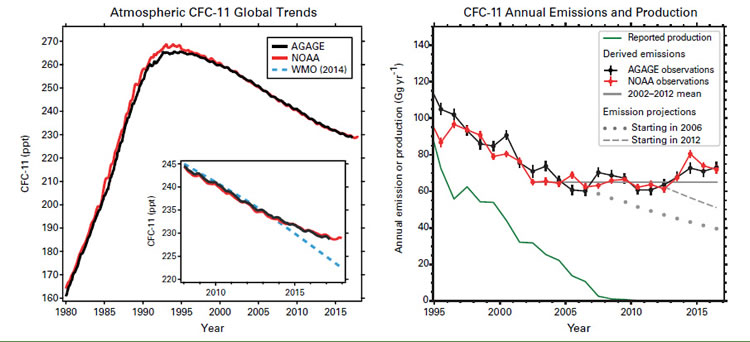
Measurements of the atmospheric abundance of the chlorofluorocarbon CFC-11, a potent greenhouse gas (GHG) and a stratospheric ozone-depleting substance (ODS) regulated under the Montreal Protocol on Substances that Deplete the Ozone Layer, show that since 2012 its rate of decline has slowed to roughly two thirds of its rate of decline during the preceding decade [1, 2]. The most likely cause of this slowing is increased emissions associated with production of CFC-11 in eastern Asia. This discovery illustrates the importance of long-term measurements of atmospheric composition, such as are carried out under the auspices of the Global Atmosphere Watch (GAW) Programme of WMO, in providing effective support and additional constraints for emissions-control legislation.
The Montreal Protocol was designed to protect the stratospheric ozone layer by restricting the production of ODSs such as CFCs. As a consequence, CFC-11 (trichlorofluoromethane, or CCl3F) production reported under the Montreal Protocol declined to zero by 2010. As CFC-11 was phased out, its atmospheric abundance peaked in the early 1990s and then declined in a manner largely consistent with declining production combined with residual emissions of CFC-11 gradually escaping from stored “banks” in existing products and equipment.
Atmospheric measurements of CFC-11 made by independent global networks show that since 2012 the rate of decrease in atmospheric CFC-11 has slowed to roughly two thirds of the rate that was observed between 2002 and 2012 [1, 2]. These global trends are shown in the left graph of the figure forthe Advanced Global Atmospheric Gases Experiment (AGAGE; shown in black) and the National Oceanic and Atmospheric Administration (NOAA; shown in red) measurement networks. Also shown in the inset to this graph is the trend that was predicted in 2014 by WMO (blue dashed) assuming adherence to the Montreal Protocol [3].
Modelling results lead to the robust conclusion that these changes are predominately related to increased CFC-11 emissions rather than to other possible causes such as changing atmospheric transport. This conclusion is supported by recent increases in the northern to southern hemisphere difference in atmospheric concentration levels. Correlations between elevated abundances of CFC-11 and other measured gases further suggest that these increases originate from emissions in eastern Asia [1].
Separate CFC-11 emission trends resulting from model calculations taken from the 2018 WMO ozone assessment [2], based on data from each of the global measurement networks AGAGE (black) and NOAA (red), are shown in the graph on the right of the figure. They are contrasted to CFC-11 production as reported under the Montreal Protocol (green). These results show a levelling off of CFC-11 emissions around 2005, followed by an emission increase of about 15% after 2012. Emission scenario projections for the years 2006 and 2012 based on atmospheric data, reported production and releases from banks are shown as dots and dashes (grey), respectively.
This work demonstrates the importance of long-term measurements of atmospheric composition, such as are carried out under the auspices of the GAW Programme, in providing observation-based information to support national emission inventories, especially in the context of agreements to address anthropogenic climate change, as well as for the recovery of the stratospheric ozone layer.
|
Table 1 provides globally averaged atmospheric abundances of the three major LLGHGs in 2017 and changes in their abundances since 2016 and 1750. Data from mobile stations (blue triangles and orange diamonds in Figure 2),with the exception of NOAA sampling in the eastern Pacific, are not used for this global analysis.
Number of stations used for the calculation of the global averages.
| CO2 |
129 |
| CH4 |
126 |
| N2O |
96 |
| CFC-11 |
23 |
| CFC-12 |
25 |
| CFC-113 |
21 |
| CCl4 |
21 |
| CH3CCl3 |
24 |
| HCFC-141b |
9 |
| HCFC-142b |
14 |
| HCFC-22 |
13 |
| HFC-134a |
10 |
| HFC-152a |
9 |
| SF6 |
85 |
|
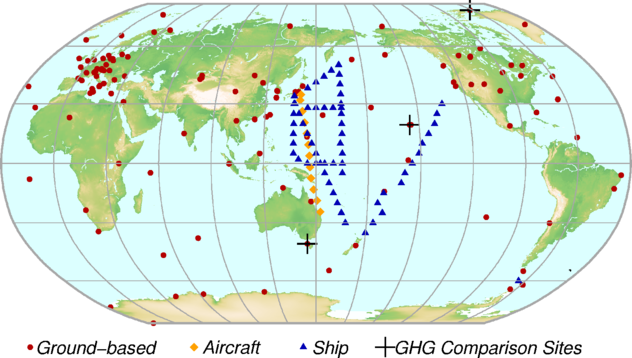
Figure 2. Figure 2. The GAW global network for CO2 in the last decade. The network for CH4 is similar.
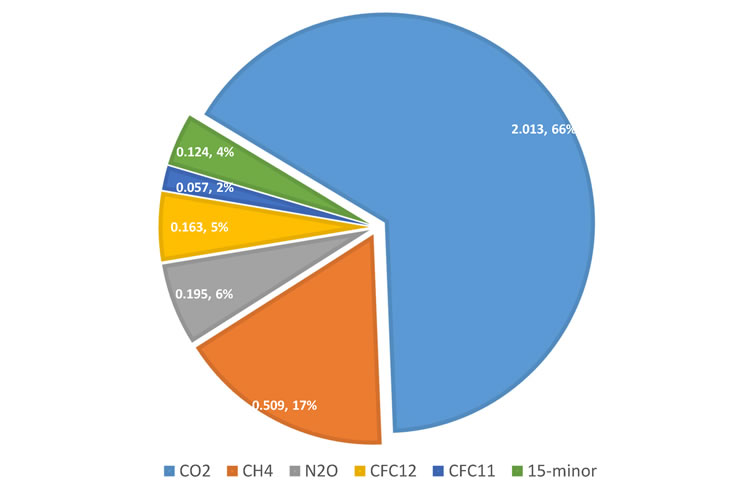
Figure 3. Increase in 2017 in global radiative forcing since pre-industrial times resulting from increased atmospheric burden of the most important LLGHGs, expressed in W per squire meter and relative to the total increase from all GHGs of 3.062 W m-2 [4]. |
The three GHGs shown in Table 1 are closely linked to anthropogenic activities and also interact strongly with the biosphere and the oceans. Predicting the evolution of the atmospheric content of GHGs requires quantitative understanding of their many sources, sinks and chemical transformations in the atmosphere. Observations from GAW provide invaluable constraints on the budgets of these and other LLGHGs, and they are used to support emission inventories preparation and evaluate satellite retrievals of LLGHG column averages. The Integrated Global Greenhouse Gas Information System (IG3IS), promoted by WMO, provides further insights on the sources of GHGs on the national and sub-national level. Some examples of the information that is delivered by the IG3IS projects can be found in the central insert of this Bulletin.
The NOAA AGGI [4] in 2017 was 1.41, representing a 41% increase in total radiative forcing by all LLGHGs since 1990 and a 1.6% increase from 2016 to 2017 (Figure 1). The total radiative forcing by all LLGHGs in 2017 (3.062 W m-2) corresponds to an equivalent CO2 mole fraction of 493 ppm [4]. Relative contributions of the other gases in the total radiative forcing since pre-industrial time are presented in Figure 3.
Table 1. Global annual surface mean abundances (2017) and trends of key GHGs from the WMO GAW global GHG observational network. Units are dry-air mole fractions, uncertainties are 68% confidence limits [5], and the averaging method is described in [7]. The number of stations used for the analyses is 129 for CO2, 126 for CH4 and 96 for N2O.
|
CO2 |
CH4 |
N2O |
Global abundance in 2017 |
405.5 ± 0.1
ppm |
1 859 ± 2
ppb |
329.9 ± 0.1
ppb |
2017 abundance relative to year 1750* |
146% |
257% |
122% |
2016–2017 absolute increase |
2.2 ppm |
7 ppb |
0.9 ppb |
2016–2017 relative increase |
0.55% |
0.38% |
0.27% |
Mean annual absolute increase during last 10 years |
2.24 ppm per year |
6.9 ppb per year |
0.93 ppb per year |
*
Assuming a pre-industrial mole fraction of 278 ppm for CO2, 722 ppb for CH4 and 270 ppb for N2O.
ATMOSPHERIC OBSERVATIONS AND ANALYSIS IN SUPPORT OF GHG EMISSION MITIGATION – EXAMPLE PROJECTS OF THE GAW IG3IS PROGRAMME
1. Atmospheric measurements reveal strong forest carbon sink in New Zealand
By Sara Mikaloff-Fletcher (National Institute of Water and Atmospheric Research Ltd, New Zealand) and Jocelyn Turnbull (GNS Science, New Zealand) |
(click to enlarge) |
(click to enlarge) |
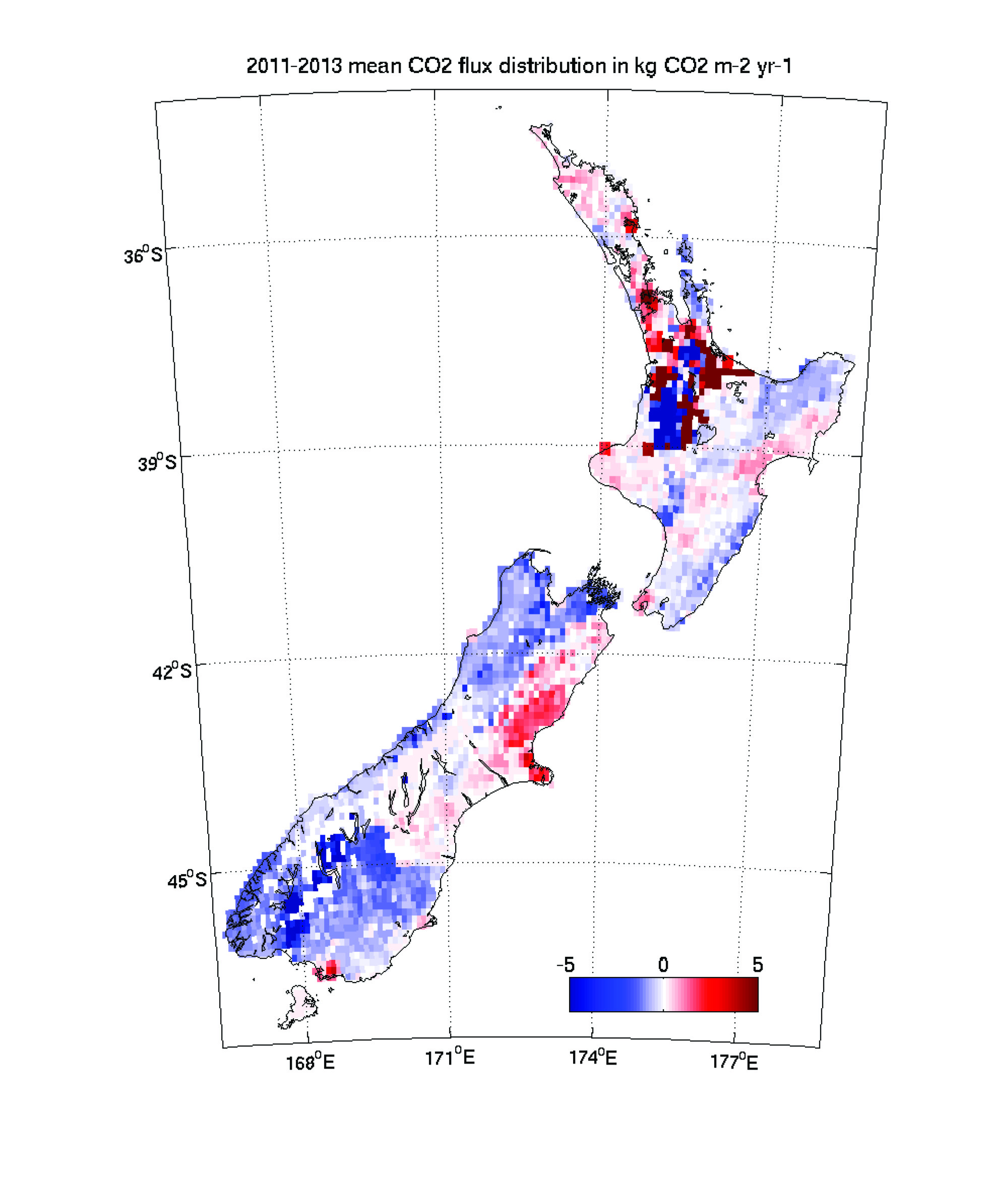
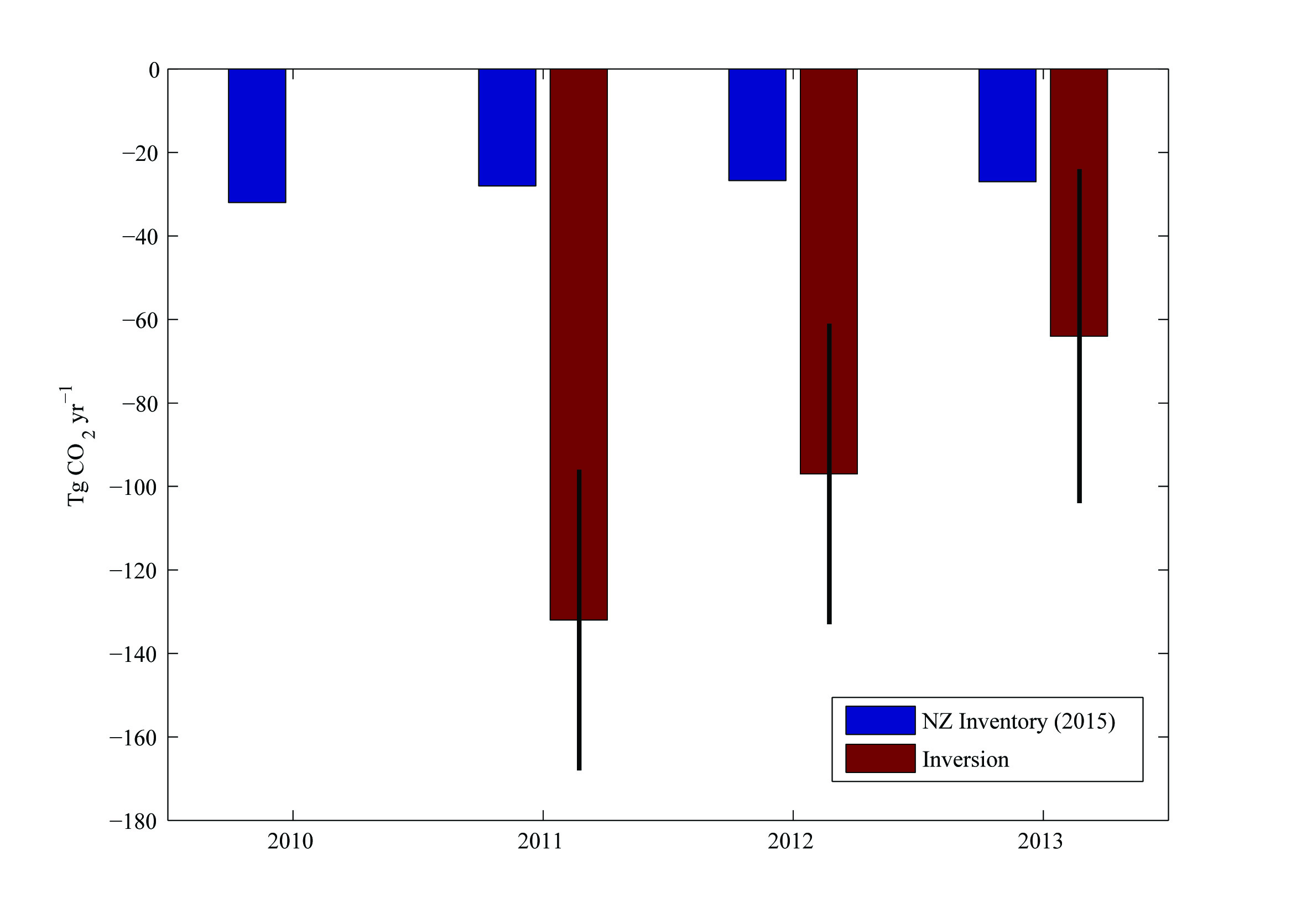
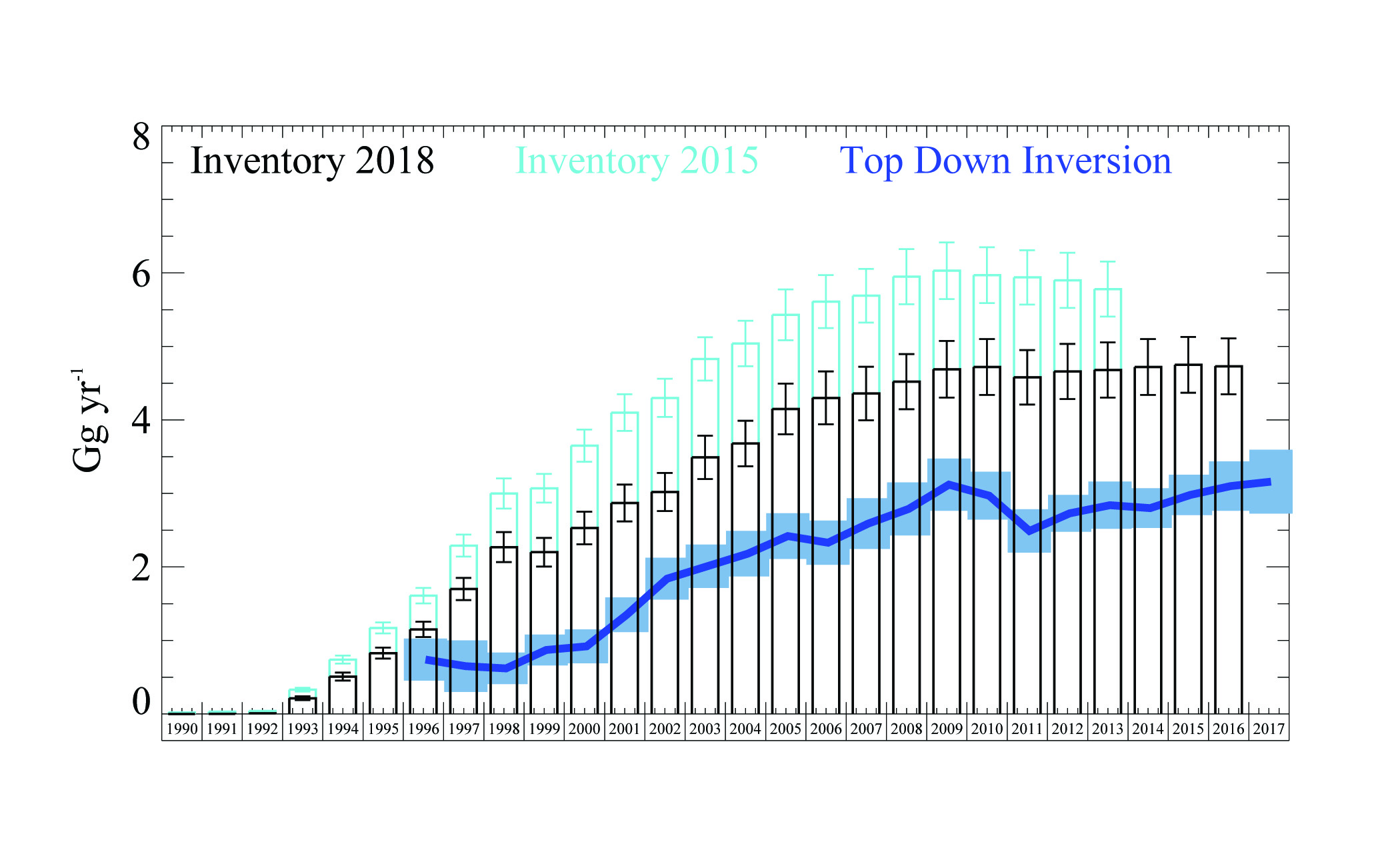
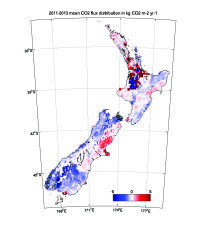
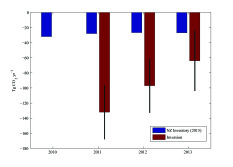
Figure 8. Geographic distribution of land-to-air CO2 flux, averaged over 2011–2013 [10]. Blue and red regions indicate net carbon uptake and release, respectively. Per-area ocean fluxes are too small to show on this scale. Fossil fuel emissions are included and reach up to 20 kg CO2 m-2 yr-1 in a few grid cells (Auckland area). The colour scale is capped to focus on natural fluxes. Inset: annual mean inverse model results [12] compared to the National Greenhouse Gas Inventory Report.
Net CO2 uptake from land use, land-use change and forestry currently offsets approximately 30% of New Zealand’s GHG emissions [10]. These land carbon sinks played a key role in meeting New Zealand’s past GHG emission targets under the United Nations Framework Convention on Climate Change (UNFCCC), and they are expected to be a major component of the nation’s strategy for future GHG mitigation. New Zealand’s National Inventory Report (NIR) estimates forest carbon uptake based on tree diameter and height measurements at a national network of study sites, and allometric equations that infer carbon mass from these measurements. This approach, which is required by current Intergovernmental Panel on Climate Change (IPCC) guidelines [11], has substantial uncertainty.
Atmospheric CO2 observations and inverse model simulations [12], illustrated in Figure 8, suggest that New Zealand’s forest carbon sink may far exceed estimates from the NIR [10] and land process models [12]. Furthermore, the atmospheric observations reveal significant interannual variability that is not detected by the NIR methodology. This study combined in situ observations of atmospheric CO2 at a network of sites with a high-resolution atmospheric model. The spatial pattern of the sink suggests that much of this missing carbon uptake occurs in Fiordland, a high rainfall region dominated by indigenous forests. The research team of New Zealand is launching a new research programme to further evaluate the processes that drive this sink. Through close engagement with users in the carbon accounting, land management and policy communities, this nationally funded programme will support the IG3IS mission to provide a bridge between science and policy for GHG monitoring and emission estimation.
2. Use of atmospheric observations of greenhouse gases to inform the United Kingdom national inventory
By Alistair Manning (UK Met Office)
To support the emission estimates that follow the IPCC protocol (“bottom-up”) [11] and are reported annually to UNFCCC, the United Kingdom uses a completely independent method (“top-down”) [13] for informing on its GHG emission estimates. The method uses a combination of atmospheric observations and modelling, and the results are also reported annually in the United Kingdom National Inventory Report to UNFCCC. Significant differences in the emissions estimated utilizing the two approaches are used by the United Kingdom Government Department of Business, Energy and Industrial Strategy (BEIS) to identify areas worthy of further investigation.
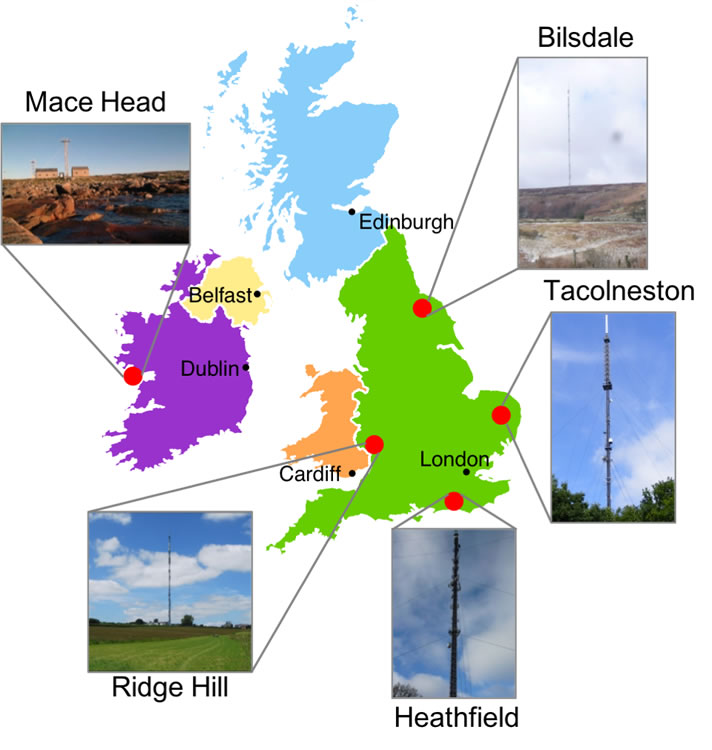
Figure 9. United Kingdom-funded DECC network of observation sites |
(click to enlarge)
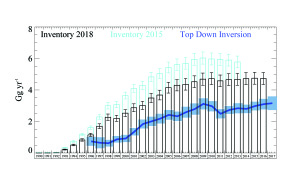
Figure 10. United Kingdom emission estimates of HFC-134a. Inventory estimates from two reporting years compared against top-down (InTEM) estimates. |
In 2012, BEIS invested in a network of observation sites (Figure 9) called the UK Deriving Emissions related to Climate Change (DECC) network [14]. These are primarily tall-tower telecommunication masts equipped with state-of-the-art observation equipment measuring CO2, CH4, N2O, HFCs, perfluorocarbons, SF6 and nitrogen trifluoride (NF3) to high precision and quality.
A recent example of how the top-down approach has been used to inform the bottom-up estimate is demonstrated in Figure 10. The country’s 2015 bottom-up estimate for HFC-134a is shown in the figure as the light blue bars with an estimated uncertainty of 8%. The top-down estimate was consistently approximately 50% of this value throughout the time series from 1994, when the observations started, until 2013, the last year of that inventory. This result, and the subsequent work undertaken [15], motivated BEIS to investigate this further and an industry expert partly revised the United Kingdom HFC-134a inventory estimates.
The result of the revised bottom-up estimate is shown in Figure 10 as black bars – it has moved to be considerably nearer to the top-down estimates. The remaining discrepancy is believed to arise from the use of assumption on a refill rate.
3. Oil and gas methane emissions in Alberta, Canada: Collecting policy-relevant atmospheric data
By Daniel Zavala-Araiza (Environmental Defense Fund, United States of America)
The oil and gas sector in Canada accounts for roughly half of total CH4 emissions in the national inventory [16]. The federal government recently announced regulations that support the goal of a 40–45% reduction of CH4 emissions from the oil and gas system below 2012 levels by the year 2025 [17].
For emission reduction goals and policies to be realistically achieved, knowledge of the current emissions baseline as well as the characteristics of the major emitting sources is a necessary condition. Therefore, a multi-scale campaign, targeting oil and gas production regions in Alberta was conducted in the fall of 2016 [18–20] and the data were used to estimate the emissions (mass balance approach). These top-down estimates were then compared with spatially explicit, region-specific inventories and industry-reported emissions. In addition, ground-based mobile (downwind, site-wide characterization using dual tracer release and Gaussian dispersion modelling) measurements allowed the characterization of the emission distributions and major sources of emissions (see Figure 11a).
In the Lloydminster region of Alberta, the major source of emissions is related to direct venting of methane to the atmosphere from the production casing. The results based on atmospheric observations suggest that emissions are three to five times greater than inventories. This large discrepancy is particularly relevant in the context of proposed regulations and emission reduction policies in Canada. If these results are conservatively extrapolated to the larger population of similar sites in Alberta, actual methane emissions from oil and gas production in the province are likely to be 25–50% higher as illustrated in Figure 11b.
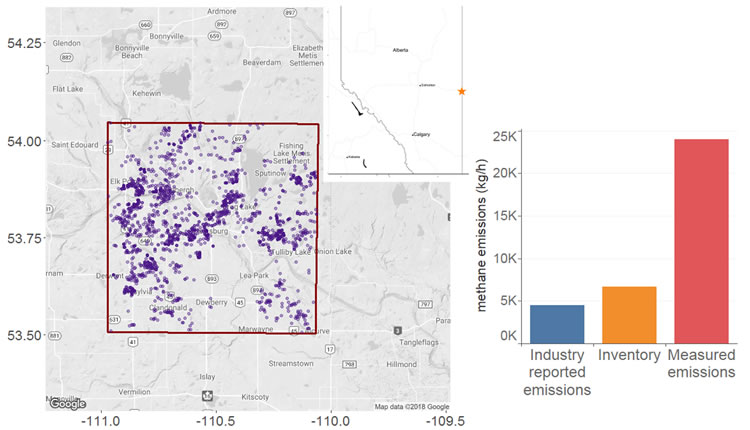
Figure 11. (a) Sampling region near Lloydminster, Alberta. The red box illustrates the source envelope where the aircraft took measurements. Purple dots inside the box represent active oil wells. (b) Comparison between measured CH4 emissions and “bottom-up” estimates based on inventory and industry reports. |
Carbon Dioxide (CO2)
Carbon dioxide is the single most important anthropogenic GHG in the atmosphere, contributing approximately 66% of the radiative forcing by LLGHGs. It is responsible for approximately 82% of the increase in radiative forcing over the past decade and over the past five years. The pre‑industrial level of 278 ppm represented a balance of fluxes among the atmosphere, the oceans and the land biosphere. Atmospheric CO2 reached 146% of the pre-industrial level in 2017, primarily because of emissions from combustion of fossil fuels and cement production (the sum of CO2 emissions was 9.9 ± 0.5 PgC(5) in 2016 [8]), deforestation and other land-use change (1.3 ± 0.7 PgC average for 2007–2016). Of the total emissions from human activities during the period 2007–2016, approximately 44% accumulated in the atmosphere, 22% in the ocean and 28% on land; the unattributed budget imbalance is 5% [8]. The portion of CO2 emitted by fossil fuel combustion that remains in the atmosphere (airborne fraction), varies inter-annually due to the high natural variability of CO2 sinks without a confirmed global trend.
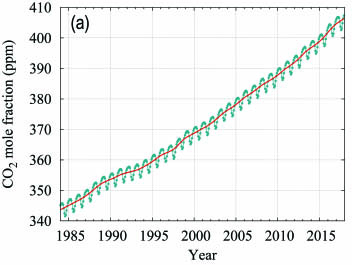
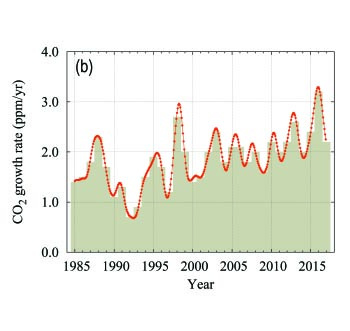
The globally averaged CO2 mole fraction in 2017 was 405.5 ± 0.1 ppm (Figure 4). The increase in annual means from 2016 to 2017, 2.2 ppm, is smaller than the increase from 2015 to 2016 (3.2 ppm) and practically equal to the average growth rate for the past decade (2.24 ppm yr-1). The higher growth rates in 2016 and 2015, in comparison with the years before 2016 and the increase from 2016 to 2017, are due in part to increased natural emissions of CO2 related to the most recent El Niño event, as explained in the twelfth edition of this Bulletin.
|
|
Figure 4. Globally averaged CO2 mole fraction (a) and its growth rate (b) from 1984 to 2017. Increases in successive annual means are shown as the shaded columns in (b). The red line in (a) is the monthly mean with the seasonal variation removed; the blue dots and line depict the monthly averages. Observations from 129 stations have been used for this analysis. |
Methane (CH4)
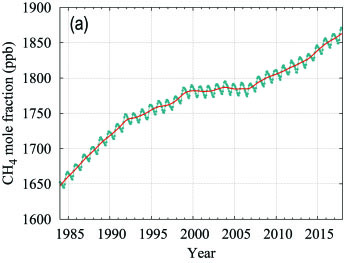
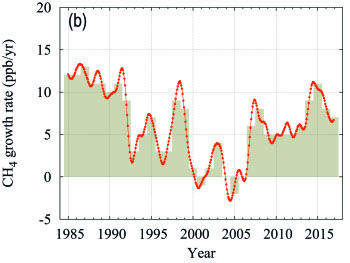
Methane contributes approximately 17% of the radiative forcing by LLGHGs. Approximately 40% of methane is emitted into the atmosphere by natural sources (e.g., wetlands and termites), and about 60% comes from anthropogenic sources (e.g., ruminants, rice agriculture, fossil fuel exploitation, landfills and biomass burning). Atmospheric CH4 reached 257% of the pre-industrial level (approximately 722 ppb) due to increased emissions from anthropogenic sources. Globally averaged CH4 calculated from in situ observations reached a new high of 1859 ± 2 ppb in 2017, an increase of 7 ppb with respect to the previous year (Figure 5). This increase is lower than the increase from 2015 to 2016 but practically equal to the average annual increase over the past decade. The mean annual increase of CH4 decreased from approximately 12 ppb yr-1 during the late 1980s to near zero during 1999–2006. Since 2007, atmospheric CH4 has been increasing again. Studies using GAW CH4 measurements indicate that increased CH4 emissions from wetlands in the tropics and from anthropogenic sources at mid-latitudes of the northern hemisphere are likely causes of this recent increase.
|
|
| Figure 5. Globally averaged CH4 mole fraction (a) and its growth rate (b) from 1984 to 2017. Increases in successive annual means are shown as the shaded columns in (b). The red line in (a) is the monthly mean with the seasonal variation removed; the blue dots and line depict the monthly averages. Observations from 126 stations have been used for this analysis. |
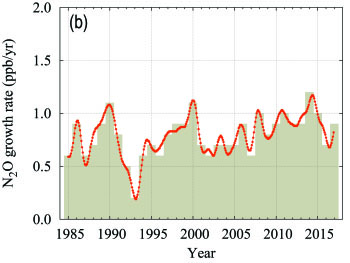
Nitrous Oxide (N2O)
|
|
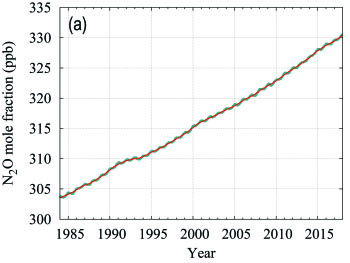
| Figure 6. Globally averaged N2O mole fraction (a) and its growth rate (b) from 1984 to 2017. Increases in successive annual means are shown as the shaded columns in (b). The red line in (a) is the monthly mean with the seasonal variation removed; in this plot it is overlapping with the blue dots and line that depict the monthly averages. Observations from 96 stations have been used for this analysis. |
Nitrous oxide contributes approximately 6% of the radiative forcing by LLGHGs. It is the third most important individual contributor to the combined forcing. N2O is emitted into the atmosphere from both natural (about 60%) and anthropogenic sources (approximately 40%), including oceans, soils, biomass burning, fertilizer use and various industrial processes. The globally averaged N2O mole fraction in 2017 reached 329.9 ± 0.1 ppb, which is 0.9 ppb above the previous year (Figure 6) and 122% of the pre-industrial level (270 ppb). The annual increase from 2016 to 2017 is higher than the increase from 2015 to 2016 and practically equal to the mean growth rate over the past 10 years (0.93 ppb yr-1). The likely causes of N2O increase in the atmosphere are an increased use of fertilizers in agriculture and increased release of N2O from soils due to an excess of atmospheric nitrogen deposition related to air pollution.
Other greenhouse gases
Sulphur hexafluoride (SF6) is a potent LLGHG. It is produced by the chemical industry, mainly as an electrical insulator in power distribution equipment. Its current mole fraction is more than twice the level observed in the mid-1990s (Figure 7a). The stratospheric ozone-depleting CFCs, together with minor halogenated gases, contribute approximately 11%(4) of the radiative forcing by LLGHGs. While CFCs and most halons are decreasing, some hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs), which are also potent GHGs, are increasing at relatively rapid rates, although they are still low in abundance (at ppt(6) levels).
|
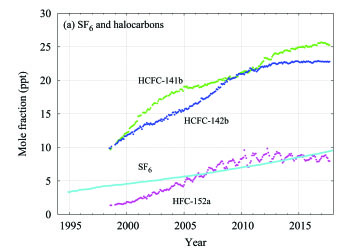
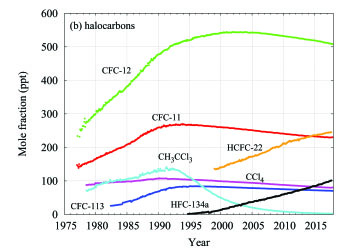
Figure 7. Monthly mean mole fractions of SF6 and the most important halocarbons: (a) SF6 and lower mole fractions of halocarbons and (b) higher halocarbon mole fractions. The numbers of stations used for the analyses are as follows: SF6 (85), CFC-11 (23), CFC-12 (25), CFC-113 (21), CCl4 (21), CH3CCl3 (24), HCFC-141b (9), HCFC-142b (14), HCFC-22 (13), HFC-134a (10), HFC-152a (9).
|
This Bulletin primarily addresses LLGHGs. Relatively short-lived tropospheric ozone [9] has a radiative forcing comparable to that of the halocarbons. Many other pollutants,such as carbon monoxide, nitrogen oxides and volatile organic compounds, although not referred to as GHGs, have small direct or indirect effects on radiative forcing. Aerosols (suspended particulate matter) are short-lived substances that alter the radiation budget. All gases mentioned herein, as well as aerosols, are monitored by the GAW Programme, with support from WMO Members and contributing networks.
Acknowledgements and links
Fifty-three WMO Members have contributed CO2 and other GHG data to WDCGG. Approximately 41% of the measurement records submitted to WDCGG were obtained at sites of the NOAA Earth System Research Laboratory cooperative air-sampling network. For other networks and stations, see GAW Report No. 229. AGAGE also contributed observations to this Bulletin. Furthermore, the GAW observational stations that contributed data to this Bulletin, shown in Figure 2, are included in the list of contributors on the WDCGG web page (https://gaw.kishou.go.jp/). They are also described in the GAW Station Information System (https://gawsis.meteoswiss.ch/) supported by MeteoSwiss.
References
[1] Montzka, S. A. et al., 2018: An unexpected and
persistent increase in global emissions of ozonedepleting
CFC 11. Nature, 557:413–417, doi:10.1038/
s41586-018-0106-2.
[2] World Meteorological Organization, 2018: Executive
Summary: Scientific Assessment of Ozone Depletion:
2018. World Meteorological Organization, Global Ozone
Research and Monitoring Project – Report No. 58.
67 pp., Geneva.
[3] ———, 2014: Scientific Assessment of Ozone Depletion:
2014. World Meteorological Organization Global Ozone
Research and Monitoring Project – Report No. 55. 416 pp.,
Geneva.
[4] Butler, J.H. and S.A. Montzka, 2018: The NOAA Annual
Greenhouse Gas Index (AGGI),http://www.esrl.noaa.
gov/gmd/aggi/aggi.html.
[5] Conway, T.J., P.P. Tans, L.S. Waterman, K.W. Thoning,
D.R. Kitzis, K.A. Masarie and N. Zhang, 1994:
Evidence for interannual variability of the carbon
cycle from the National Oceanic and Atmospheric
Administration/Climate Monitoring and Diagnostics
Laboratory Global Air Sampling Network. Journal of
Geophysical Research – Atmospheres, 99:22831–22855,
doi:10.1029/94JD01951.
[6] National Oceanic and Atmospheric Administration
Earth System Research Laboratory, 2018: Trends in
atmospheric carbon dioxide, http://www.esrl.noaa.
gov/gmd/ccgg/trends/.
[7] World Meteorological Organization, 2009: Technical
Report of Global Analysis Method for Major Greenhouse
Gases by the World Data Center for Greenhouse Gases.
(Y. Tsutsumi, K. Mori, T. Hirahara, M. Ikegami and
T.J. Conway). GAW Report No. 184 (WMO/TD-No. 1473),
Geneva, https://www.wmo.int/pages/prog/arep/gaw/documents/TD_1473_GAW184_web.pdf.
[8] Le Quéré, C. et al., 2018: Global carbon budget 2017.
Earth System Science Data, 7(10):405–448, doi:10.5194/essd-10-405-2018.
[9] World Meteorological Organization, 2018: WMO
Reactive Gases Bulletin No. 2: Highlights from the Global
Atmosphere Watch Programme, https://library.wmo.int/doc_num.php?explnum_id=5244.
[10] Ministry for the Environment – Manatu Mo
Te Taiao, 2018: New Zealand's Greenhouse Gas
Inventory 1990–2016. Publication number: ME 1351.
Wellington.
[11] Intergovernmental Panel on Climate Change,
2006: 2006 IPCC Guidelines for National Greenhouse
Gas Inventories, Volume 1, General Guidance and
Repor ting (S. Eggleston, L. Buendia, K. Miwa,
T. Ngara and K. Tanabe, eds). Prepared by the
National Greenhouse Gas Inventories Programme.
Section 6.10.2. IGES, Japan.
[12] Steinkamp, K., S.E. Mikaloff-Fletcher, G. Brailsford,
D. Smale, S. Moore, E. Keller, T. Baisden, H. Mukai and
B. Stephens, 2017: Atmospheric CO2 observations and
models suggest strong carbon uptake by indigenous
forests in New Zealand. Atmospheric Chemistry and
Physics, 17:47–76.
[13] Stanley, K. M., A. Grant, S. O’Doherty, D. Young,
A.J. Manning, A.R. Stavert, T.G. Spain, P.K. Salameh,
C.M. Harth, P.G. Simmonds, W.T. Sturges, D.E. Oram
and R.G. Derwent, 2018: Greenhouse gas measurements
from a UK network of tall towers: Technical description
and first results. Atmospheric Measurement Techniques,
11(3):1437–1458.
[14] Arnold, T., A.J. Manning, J. Kim, L. Shanlan,
H. Webster, D. Thomson, J. Mühle, R.F. Weiss, S. Park
and S. O’Doherty, 2018: Inverse modelling of CF4 and
NF3 emissions in East Asia. Atmospheric Chemistry
and Physics, 18:13305–13320.
[15] Say, D., A.J. Manning, S. O’Doherty, M. Rigby,
D. Young and A. Grant, 2016: Re-evaluation of the UK’s
HFC-134a emissions inventory based on atmospheric
observations. Environmental Science and Technology,
50(20):11129–11136.
[16] Environment and Climate Change Canada, 2016:
National Inventory Report 1990–2014: Greenhouse
Gas Sources and Sinks in Canada, http://publications.gc.ca/collections/collection_2016/eccc/En81-4-1-2014-eng.pdf.
[17] ———, 2017: Regulations respecting reduction in
the release of methane and certain volatile organic
compounds (upstream oil and gas sector). Canada
Gazette, Part I, Volume 151, http://www.gazette.gc.ca/rp-pr/p1/2017/2017-05-27/html/reg1-eng.php.
[18] Johnson, MR, D.R. Tyner, S. Conley, S. Schwietzke
and D. Zavala-Araiza, 2017: Comparisons of airborne
measurements and inventory estimates of methane
emissions in the Alberta upstream oil and gas
sector. Environmental Science and Technology, 51(21):13008–13017.
[19] Zavala-Araiza D., S.C. Herndon, J.R. Roscioli,
T.I. Yacovitch, M.R. Johnson, D.R. Tyner, M. Omara and
B. Knighton, 2018: Methane emissions from oil and gas
production sites in Alberta, Canada. Elementa Science
of the Anthropocene, 6(1):27.
[20] Roscioli J.R., S.C. Herndon, T.I. Yacovitch,
W.B. Knighton, D. Zavala-Araiza, M.R. Johnson and
D.R. Tyner, 2018: Characterization of methane emissions
from five cold heavy oil production with sands (CHOPS)
facilities. Journal of the Air and Waste Management
Association, 68(7):671–684
Terms used in this Bulletin are explained in the glossary.
|